
Total de visitas: 338086
|
Mutações
Antes de começar a desenvolver o tema convém perceber o que são afinal as mutações. Segundo o Manual de Genética Médica de Fernando J. Regateiro, entende-se por mutação toda a alteração permanente do genoma de uma célula em relação a uma sequência de DNA de referência, transmissível às células filhas; pode dizer respeito a um único nucleótido ou ser mais extensa; uma mutação que ocorre nos gâmetas é hereditária; uma mutação que ocorre nas células somáticas não é hereditária.
Existem diversos tipos de mutações, sendo importante salientar as suas causas e, a título de curiosidade, a frequência com a qual elas aparecem.
A frequência de ocorrência de uma mutação num gene não depende apenas de factores intrínsecos mas também de factores extrínsecos, considerando-se, respectivamente, mutações espontâneas e mutações induzidas. Estas últimas geralmente ocorrem com uma frequência muito mais elevada do que as mutações espontâneas. Os erros de replicação podem também levar a alterações do DNA.
Das diferentes taxas de mutação observadas para diferentes genes que têm como causa factores intrínsecos ao DNA, as mais relevantes são:
- A extenção do gene em questão (quanto mais extenso maior a probabilidade da ocorrência de uma mutação);
- A extensão e o número de intrões, pela maior probabilidade de erro durante o processamento do RNAm;
- O tipo de bases presente (quando se interrompe a ligação glicosídica entre a desoxirribose e a base, pode acontecer que nas posições ocupadas pela adenina ou pela guanina bases púricas se perca uma delas, dando origem a um lugar apurínico; da mesma forma, a probabilidade de ocorrência de mutações, devido à desaminação da forma 5metilcitosina em timina, aumenta nas regiões de DNA com maior percentagem de bases de citosina);
- A existência de sequências repetitivas (principalmente devido à possibilidade de expansão do número de tripletos presentes).
Os factores que provocam mutações e que são extrínsecos ao DNA tomam a designação de mutagéneos, onde se incluem os agentes ambientais de natureza física e química.
Dentro dos agentes de natureza física encontram-se as radiações não-ionizantes, que são capazes de fomentar ligações covalentes entre as bases pirímidicas adjacentes (por exemplo, as radiações ultra-violeta), e as radiações ionizantes associadas à quebra da dupla cadeia de DNA ou à troca de bases (por exemplo, raios X, neutrões, partículas α e β e radiação γ).
Nos agentes de natureza química encontram-se os carcinógeneos químicos capazes de promover metilação e aductos resultantes das modificações covalentes do DNA ou da acção de espécies livres de oxigénio; dos corantes de acridina e do efeito da proflavina (ambos se intercalam na cadeia nucleotídica, podendo provocar mutações frameshift termo que será esclarecido mais à frente); a incorporação de DNA de análogos das bases normalmente encontradas; moléculas mutagénicas como o formaldeído, o cloreto de vinilo, o nitrato de sódio, o benzeno, os agentes alquilantes (por exemplo, nitrosoguanidina e etilmetanosulfatonato), ou a aflatoxina B1 (que gera locais apurínicos). Há também os radicais livres resultantes dos processos biológicos e os agentes químicos muito conhecidos devido ao seu papel na história mundial como, por exemplo, o gás mostarda, as bombas nucleares, o gás sarin, entre outros.
As mutações podem ocorrer em diversos locais do DNA como, por exemplo, ao nível das regiões génicas codificadoras - os exões (estas por sua vez irão alterar o RNA mensageiro e, normalmente, as proteínas resultantes da tradução deste, a não ser que ocorram no terceiro nucleótido de cada codão, dado que aí já podem não afectar o aminoácido da proteína devido à redundância do código genético), ou ao nível das regiões não codificadoras intrões, sequências flanqueadoras em posição 5 e 3, extensas regiões intergenéticas (que, normalmente, não afectam a composição das proteínas).
Quanto aos tipos de mutações, elas podem-se dividir em dois grandes grupos: as mutações génicas e as mutações cromossómicas, dividindo-se ainda estas últimas em mutações estruturais e mutações numéricas.
Das alterações cromossómicas de natureza estrutural podem advir diversas consequências, que variam consoante a região alterada e a extensão da região em causa. A identificação da existência deste tipo de mutações beneficiou com o recurso a estudos citogenéticos com bandeamento (em particular o de alta resolução) e resultam de quebras num cromossoma com consequente rearranjo diferente. Estes rearranjos podem ser ou não equilibrados. Diz-se que se trata de um rearranjo equilibrado quando não há ganho nem perda de material cromossómico em quantidade ou qualidade que se reflicta em consequências patológicas. Por exemplo, os portadores de células germinais com este tipo de rearranjo podem originar descendência com manifestações patológicas devido a uma dosagem génica anormal (por excesso ou defeito) originando gâmetas com complemento cromossómico desequilibrado. Considera-se que se trata de um rearranjo não equilibrado quando a uma quantidade de material cromossómico anormal se associa, geralmente, um fenótipo anormal.
As mutações estruturais dos cromossomas podem ser intercromossómicas (insersões, translocações robertsonianas e translocações recíprocas) ou intracromossómicas (deleções intersticiais e terminais, isocromossoma, cromossoma em anel, inversões pericêntricas e paracêntricas).
As translocações são as alterações que ocorrem mais vulgarmente na espécie humana e consistem na recombinação ou troca de fragmentos de cromossomas não homólogos, não existindo geralmente perda de material cromossómico ou, no caso da sua ocorrência, é de tal modo que não afecta o fenótipo do indivíduo portador de uma forma equilibrada de translocação. Há três tipos de translocação: a insercional, a recíproca e a robertsoniana.
Na translocação insercional uma porção de cromossoma muda de local entre cromossomas diferentes ou dentro do mesmo cromossoma. Para que tal acontece é necessário que se verifique a existência de três pontos de quebra: um no cromossoma que, ao abrir, vai permitir a inserção do fragmento translocado, e dois num cromossoma para que o fragmento possa ser libertado. Assim e devido a isto, há um risco elevado da descendência ter anomalias, é da ordem dos 50%.
Nas translocações recíprocas ocorre o intercâmbio de dois fragmentos cromossómicos localizados em posição distal em relação à quebra ocorrida nos braços de dois cromossomas não homólogos, podendo qualquer cromossoma e qualquer dos braços estar envolvido. No indivíduo em que ocorreu esta translocação, estabelece-se um rearranjo equilibrado e, embora a morfologia dos cromossomas derivados seja diferente da dos iniciais, normalmente não há efeitos fenotípicos desde que a quebra não lese a estrutura de nenhum gene. Pode, no entanto, ocorrer que a segregação meiótica origine gâmetas com conteúdo cromossómico não equilibrado, responsável por alterações fenotípicas em descendentes. Durante o emparelhamento meiótico formam-se geralmente quadrivalentes com segregação cromossómica anormal durante a meiose, registando-se, por isso, nos indivíduos portadores desta translocação, abortos repetidos e infertilidade, podendo os descendentes ser afectados por monossomia ou trissomia parciais, às quais se associa normalmente um fenótipo com malformações congénitas múltiplas. As translocações recíprocas que são compatíveis com a vida têm um risco de recorrência de portadores raramente superior a 20% ou 30%, sendo habitualmente menor. Este risco de recorrência é ainda mais baixo quando as alterações são extensas, devido à morte do embrião ou do feto e ao aborto subsequente.

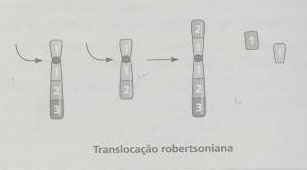
Na translocação robertsoniana, a quebra dá-se no centrómero ou próxima das sequências repetitivas do braço curto. Os braços longos dos dois cromossomas unem-se pelos topos originados pela quebra originando uma nova forma de cromossoma, e os fragmentos acêntricos correspondentes aos braços curtos perdem-se em subsequentes divisões celulares (o cariótipo, numa situação equilibrada, será 45, XX ou XY). O portador de uma translocação equilibrada apresenta um fenótipo normal pois nos braços curtos apenas se localizam genes ribossomais (cuja falta não se faz sentir, dado que os outros acrocêntricos também possuem este tipo de genes) e heterocromatina constitutiva. Todos os gâmetas produzidos por indivíduos com translocação robertsoniana equilibrada entre cromossomas acrocêntricos homólogos são cromossomicamente anormais.
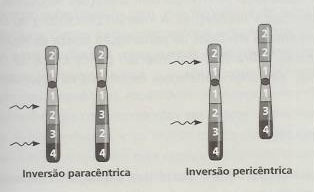
As inversões cromossómicas podem ser resultado de neo-mutações ou serem herdadas ao longo de sucessivas gerações de uma família, e são relativamente frequentes (calcula-se que cerca de um em cada 1000 indivíduos seja afectado). Nestas não ocorre perda de material cromossómico, ocorrendo quando se produzem duas quebras num cromossoma seguidas de rotação de 180º do fragmento delimitado pelas quebras, alterando-se, consequentemente, a ordem dos genes no cromossoma. Como já foi referido, as inversões podem ser pericêntricas (mais frequentes) ou paracêntricas. Nas primeiras, dá-se uma quebra em cada braço de um cromossoma, ficando o centrómero incluído no fragmento sujeito a inversão (é por isso habitual observar-se uma alteração morfológica bem aparente, inclusive da posição do centrómero). Nas inversões paracêntricas as duas quebras dão-se no mesmo braço do cromossoma, não implicando desta forma a alteração da posição do centrómero devido ao rearranjo cromossomático, nem a morfologia do cromossoma. Apesar das inversões não se encontrarem associadas a alterações patológicas significativas para os seus portadores, o risco de alterações cromossómicas na descendência é elevado, devido à produção de gâmetas com desequilíbrio cromossómico. Dado que o emparelhamento dos homólogos durante a profase I da meiose implica um alinhamento perfeito entre segmentos homólogos, é necessário que ocorra looping da região cromossómica com a inversão. Desta forma, nas inversões paracêntricas, se ocorrer crossing-over podem ser gerados cromossomas com recombinações instáveis sob a forma de acêntricos curtos que se perdem em divisões celulares subsequentes ou de dicêntricos com duplicações e deleções. Normalmente, na descendência, só se encontram cromossomas normais ou com inversão equilibrada, originando-se crianças normais, dada a instabilidade das formas cromossómicas anormais. Nas inversões pericêntricas forma-se igualmente looping, mas o crossing-over leva à produção de cromossomas com deleções e duplicações nas regiões distais em relação aos dois pontos de quebra. Dependente do tamanho destas duplicações, deleções e do cromossoma envolvido estão os efeitos fenotípicos verificáveis na descendência. Naturalmente que quanto mais próximos dos telómeros se encontrarem os pontos de quebra, menor é a extensão das alterações cromossómicas e, por isso, maior a probabilidade de sobrevivência do feto. Devido a deleções e duplicações extensas pode acontecer que, num casal em que um dos membros tem uma inversão pericêntrica, se dêem abortos espontâneos ou que nasçam crianças com alterações fenotípicas associadas a pequenas deleções e duplicações. A título de curiosidade, posso referir que a inversão pericêntrica do cromossoma 1 está associada a casos de perturbação severa da espermatogénese, e que o mesmo tipo de inversão no cromossoma 9 não origina alterações fenotípicas na descendência.
As duplicações consistem na existência de duas cópias de um segmento de um cromossoma. Se a estas duas cópias se adicionar a cópia de outro cromossoma homólogo verifica-se que uma duplicação origina uma trissomia parcial. Também da extensão do material cromossómico que se encontra envolvido na duplicação (no que se refere ao número de cópias e ao número de genes), estão dependentes os efeitos fenotípicos que se irão verificar. As duplicações parciais têm, consequências menos graves do que as deleções parciais. As duplicações cromossómicas podem ocorrer na descendência de um indivíduo com uma translocação recíproca. Pela segregação meiótica, um gâmeta pode herdar duas regiões cromossómicas homólogas, uma no cromossoma adequado e na posição original e a região homóloga supranumerária sujeita a translocação e veiculada pelo cromossoma hospedeiro. Também o crossing-over assimétrico durante a meiose pode originar duplicação. O risco de recorrência de duplicações é baixo, a não ser que, num dos progenitores, esteja presente um rearranjo (translocação ou inversão).

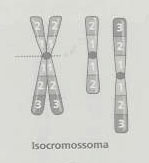
No isocromossoma o material dos dois braços tem uma constituição igual (como uma imagem em espelho a partir do centrómero), e o outro braço perde-se. Um dos mecanismos que está na origem dos isocromossomas é a divisão transversal do centrómero na meiose ou na mitose, separando as duas cópias dos braços longos para um lado e as duas cópias dos braços longos para outro. Também podem ser originados através da translocação robertsoniana entre os braços longos de cromossomas acrocêntricos homólogos. O isocromossoma dos autossomas não acrocêntricos é letal, por causa da extensa deleção de material que origina (todo o material de um dos braços). Por outro lado, na condição equilibrada, o isocromossoma dos braços longos dos cromossomas acrocêntricos não provoca alterações fenotípicas visíveis. O isocromossoma X é compatível com a vida, sendo mais frequente o isocromossoma do braço longo do cromossoma X associado ao síndrome de Turner (45,X0). Os descendentes de um progenitor com isocromossoma dos braços longos de um autossoma acrocêntrico, têm um risco de trissomia igual a 100%. Todos os gâmetas produzidos são nulissómicos (conduzindo a uma monomia autossómica, o que é letal) ou dissómicos (conduzindo a uma trissomia).
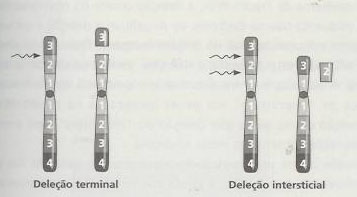
O estudo das deleções foi muito útil para a localização dos genes. Estas consistem na perda de um fragmento acêntrico de um cromossoma, o que origina uma condição de monossomia parcial. As deleções podem ser intersticiais (quando ocorrem duas quebras num cromossoma e se perde o fragmento cromossómico localizado entre as quebras) ou terminais (quando ocorre quebra do cromossoma num único ponto).
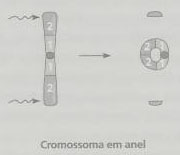
Pode acontecer também que se verifique uma deleção num cromossoma e uma duplicação no outro em consequência de crossing-over assimétrico, por exemplo. Quando ocorre, em cada um dos braços de um cromossoma, uma quebra cromossómica que envolve a perda dos telómeros, pode-se verificar a fusão dos topos adesivos, originando-se um cromossoma em anel. O risco de recorrência de uma deleção é negligenciável, a não ser que se verifique um rearranjo cromossómico num dos progenitores. Devido a isto, caso se constate que um descendente tem esta alteração estrutural, deve-se efectuar um cariótipo dos progenitores, de modo a ser realizado o aconselhamento genético.
Quanto às alterações cromossómicas numéricas importa começar por referir que a presença de um complemento cromossómico normal (correspondente no caso da espécie humana a 46 cromossomas, ou seja, um número diplóide (2n) de cromossomas) denomina-se por euploidia.
Quando duas ou mais linhas celulares provenientes de um mesmo zigoto estão presentes num indivíduo, diz-se que se trata de um mosaico. Neste caso, embora epigeneticamente modificado, o genótipo pode ser o mesmo, como acontece no sexo feminino com os dois cromossomas X, após a lionização . No entanto, o cariótipo ou o genótipo pode ser diferente, como resultado de mutação ou não-disjunção cromossómica.
De ocorrência muito rara são os casos em que num indivíduo se encontram células provenientes de dois zigotos diferentes, designando-se o indivíduo por quimera. Pode eventualmente acontecer que haja uma diferenciação sexual anormal devido à presença de uma linha celular 46,XY e outra 46,XX.
Na dissomia uniparental, um dos pares de cromossomas homólogos de um complemento diplóide tem origem num mesmo progenitor.
Quando se verifica uma alteração numérica em que o número de cromossomas é múltiplo de n, mas diferente de 2, trata-se de uma poliploidia (pode ser uma triploidia (3n) ou uma tetraploidia (4n), por exemplo). Os zigotos poliplóides sucumbem precocemente, na fase embrionária ou fetal. Se eventualmente o feto se desenvolver até ao período peri-natal, ocorrerá a sua morte após alguns dias de sobrevivência, apresentando anomalias na generalidade dos órgãos. A maioria das triploidias ocorre devido à fecundação por dois espermatozóides haplóides num ovócito, e são responsáveis por quase um sexto de todos os abortos espontâneos. Pode também ser resultado da fecundação de um ovócito diplóide por ausência de redução meiótica, da não eliminação de um globo polar ou da fecundação do ovócito por um espermatozóide diplóide.
Designam-se de aneuploidias as alterações em que o número de cromossomas difere de 2n por um ou mais cromossomas, sem ser múltiplo de n, e podem ser: monossomia (quando há falta de um cromossoma), trissomia (quando há um cromossoma a mais), tetrassomia (quando há dois cromossomas excedentes). Se a diminuição ou aumento do material cromossómico envolver apenas uma parte de um cromossoma designa-se, respectivamente, monossomia ou trissomia parcial. As aneuploidias são responsáveis por cerca de 50% dos abortos espontâneos, e podem resultar da não-disjunção meiótica ou mitótica, ou de anomalias da mitose (como o atraso da migração de um cromossoma para o pólo do fuso acromático.
As monossomias completas originam alterações de tal gravidade nos embriões que param o seu desenvolvimento. Apesar de haver referência a casos excepcionais de sobrevivência de indivíduos com monossomia 21, a monossomia 45,X (responsável pela Síndroma de Turner) é a única compatível com a vida. As observações comparativas entre as alterações que envolvem a perda de material cromossómico com o seu ganho, indicam-nos que as consequências mais graves advêm da perda de material (compare-se, por exemplo, as consequências de uma monossomia com uma trissomia).
As aneuploidias heterossómicas são menos severas do que as aneuploidias autossómicas. Teoricamente, podem ocorrer como 45,X, 45,Y, 47,XXX, 47,XXY, 47,XYY. A monossomia 45,Y não é compatível com a vida dada a ausência dos genes presentes no cromossoma X. A monossomia 45,X é compatível com a vida dado que, funcionalmente, faltam apenas os genes correspondentes aos que não são inactivados num dos cromossoma X de um cariótipo normal 46,XX. A trissomia 47,XXY pode ter origem na não-disjunção ocorrida na primeira divisão da espermatogénese, ou na ocorrida na primeira ou segunda divisão da oogénese. A trissomia 47,XYY resulta também da não-disjunção mas desta feita ocorrida na segunda divisão da meiose paterna. A trissomia 47,XXX e a trissomia 47,XXY (síndroma de Klinefelter) têm consequências menos severas do que as restantes devido à lionização, ao reduzir as consequências que resultariam do efeito de dosagem génica provocado pelo excesso de informação devido ao aumento do número de cromossomas X. podem ainda encontrar-se polissomias do cromossoma X (48,XXXY; 48,XXXX; 49, XXXXY) condições em que ocorre igualmente lionização dos cromossomas X supranumerários. Na síndroma 47,XYY é a reduzida dimensão dos cromossomas Y e o reduzido número de genes que estes comportam que limitam os efeitos da dosagem génica.
Também as mutações génicas podem ser de vários tipos: silenciosas (ou sinónimas), missense, nonsense, frameshift, deleções e ainda inserções (mutações dinâmicas). As mutações silenciosas, missense e as nonsense são mutações pontuais pois resultam da substituição de uma única base. As mutações silenciosas são responsáveis por 25% das mutações pontuais, que resultam da substituição de uma base nucleotídica, em posição que não altera o aminoácido em questão, embora altere o codão (o que se torna possível dada a redundância do código genético). No entanto, a presença de um codão alternativo para um mesmo aminoácido pode interferir com a precisão e a precisão da transcrição e, posteriormente, da tradução, devido à reduzida disponibilidade de moléculas de RNA transferência específicas.
As mutações missense ocorrem por substituição de uma base nucleotídica o que vai fazer com que o codão resultante codifique um aminoácido diferente. As consequências patogénicas deste tipo de mutação pontual podem ser graves ou moderadas, dependendo da intensidade com que afecta a actividade funcional da proteína que o gene codifica. Um dos exemplos deste tipo de mutação é a alteração do sexto codão do RNA mensageiro produzido a partir do gene da globina β mutado (de GAG para GUG), provocando a substituição do aminoácido glutamina por valina, o que tem um efeito patogénico que se reflecte na drepanocitose (ou anemia falciforme). Neste caso, a alteração de apenas um aminoácido altera a hemoglobina normal (Hb A) para hemoglobina S (Hb S).
As mutações nonsense resultam de alterações pontuais do DNA que convertem um codão que codifica para um aminoácido em codão stop no RNA (UAA, UAG, UGA). Esta designação deve-se ao facto do codão não especificar para nenhum aminoácido. Aquando da tradução do RNAm, a presença de um codão stop em posição anormal, gera um sinal que os mecanismos de tradução interpretam como se estivesse completa a tradução da proteína. Assim, forma-se uma proteína truncada por paragem prematura da tradução. Se a mutação for proximal (em relação ao extremo 5 do gene), o polipéptido traduzido não terá actividade funcional e será rapidamente degradado. Se for mais distal (próxima do extremo 3) pode ser relativamente estável e ter actividade funcional. Pode também acontecer que uma mutação altere um codão stop para um codão que codifica um aminoácido, formando-se neste caso um polipéptido mais longo do que o normal, durante a tradução do RNAm. Um exemplo é dado pela produção de hemoglobina Constant Spring, na qual uma mutação do gene da globina α transforma o codão stop UAA (da posição 142) em codão CAA, que codifica a glutamina. Em resultado disto, a globina α é mais longa e os portadores desenvolvem talassémia α.
As mutações dinâmicas consistem na expansão do número de unidades repetitivas tipicamente constituídas por tripletos presentes num determinado gene ou na sua vizinhança. Em condições normais, um indivíduo é portador de um reduzido número de tripletos repetidos sequencialmente. A expansão repetitiva do número de tripletos presentes no progenitor ocorre durante a meiose, durante as fases precoces do desenvolvimento fetal devido à instabilidade mitótica pós-zigótica, ou ainda durante as duas fases. A transmissão das expansões correspondentes a este tipo de mutação pode ocorrer por via materna e paterna como na distrofia miotónica, apenas por via paterna como ocorre quase sempre na forma juvenil da coreia de Huntington ou apenas por via materna como na síndroma do X-frágil. A expansão não afecta a expressão normal do fenótipo, até um certo número de unidades repetitivas, denominando-se esta fase de pré-mutação. O número de unidades repetitivas não se mantém constante durante o processo de transmissão às gerações, podendo diminuir ou aumentar. A partir de um certo número de tripletos, variável de acordo com as doenças, observa-se um efeito patogénico em relação com essa expansão.
As deleções ou inserções de um ou mais codões não alteram a grelha de leitura do RNAm, mas originam proteínas que se encontram em excesso, ou que faltam, em um ou mais aminoácidos, consoante o caso. As consequências destas alterações prendem-se com a localização e com a intensidade com que afectam a função da proteína. Quando dizem respeito a um número de nucleótidos que não é múltiplo de três, a mutação origina uma alteração da grelha de leitura (mutação frameshift ver tabela). Deste modo, a reorganização da sequência de nucleótidos do DNA codificador em tripletos vai dar origem a uma nova sequência de codões diferentes, sem relação com a sequência de codões original. Consequentemente, a composição em aminoácidos da proteína codificada é diferente, a partir do local em que ocorreu a mutação, sem relação com a sequência codificadora original. É frequente verificar-se que o novo reagrupamento dos nucleótidos em codões origina, a breve trecho, um codão stop que vai interromper prematuramente a formação da proteína. É também de referir que as sequências de DNA causadoras de mutações frameshift por inserção podem resultar de transposões (jumping genes). Estes elementos móveis do genoma têm a capacidade para se replicarem e para produzirem múltiplas cópias de si mesmos. Estas podem posteriormente inserir-se noutros locais do genoma. Se a sequência inserida não for um múltiplo de três, vai alterar a grelha de leitura. Podem, por outro lado, interferir com a transcrição do gene em que se inserem. Entre as situações de doença atribuídas à inserção de transposões encontram-se os casos familiares de cancro da mama, nos casos de hemofilia e de polipose cólica, por exemplo.

Consequências das Mutações
A caracterização da patogenicidade de uma mutação é demonstrada quando é rara na população geral (inferior a 1%), quando tem um efeito na proteína funcional, ou quando segrega com a doença em famílias.
A gravidade das consequências das mutações depende das alterações que provocam a nível da capacidade funcional da proteína que codificam (em relação com o tipo de aminoácido que é substituído e com o local do gene em que ocorrem) e do impacto dessa modificação do organismo no seu todo. Algumas mutações não acarretam grandes problemas (como o daltonismo, por exemplo), mas outras geram doenças graves (como, por exemplo, fenilcetonúria, fibrose quística, distrofia muscular de Duchenne, coreia de Huntington, e algumas formas de cancro). Pode acontecer também que mutações diferentes num mesmo gene originem fenótipos patológicos diferentes (por exemplo, as mutações no gene da distrofia podem ter como consequência uma distrofia muscular de Duchenne ou uma distrofia muscular de Becker). Uma mutação pode ainda afectar apenas uma função ou, por outro lado, alterar diversas estruturas ou funções do organismo, o que se traduz num efeito pleiotrópico .
No que toca aos efeitos das mutações, estas podem ser benéficas, neutras ou deletérias. Nestas últimas os efeitos a nível individual e de descendência têm a ver com a expressividade e com a penetrância (incompleta/completa, tardia/precoce). Estas mutações estão sujeitas à pressão da selecção, sendo eliminadas da espécie as que geram desvantagem biológica. Já nas mutações neutras o portador não é afectado, e nas mutações benéficas há vantagem para o seu portador, directamente em termos reprodutivos ou por melhor adaptação ao meio. Desta forma, os alelos que concedem vantagem tendem a ser mais frequentes nas populações em que este efeito se observa.
Uma mutação por deleção de uma base, aparentemente neutra, como a que se verifica no gene que codifica o receptor CCR5 presente na superfície dos linfócitos T helper, passa a ser um benefício de extrema relevância para os indivíduos portadores da mutação que sejam infectados pelo retrovítus do HIV. Para que estes infectem os linfócitos precisam de se ligar ao receptor já referido e também ao receptor de membrana CD4. Quando a mutação se encontra presente em homozigotia, as moléculas CCR5 não migram para a superfície das células, razão pela qual não se dá a infecção dos linfócitos, ao contrário do que acontece quando as moléculas de CCR5 são normais. Nesta condição, os indivíduos deficientes têm vantagem quando comparados com os indivíduos ditos normais. Assim, na ausência de infecção, não há comprometimento das defesas imunológicas e os indivíduos, ainda que seropositivos, não desenvolvem SIDA (Síndroma de Imunodeficiência Humana Adquirida), pelo que não são doentes. Nos heterozigóticos verifica-se um desenvolvimento mais lento da SIDA, comparativamente com os homozigóticos para o alelo normal.
Entre os mecanismos que são responsáveis pela enorme diversidade humana podemos encontrar as mutações neutras e as benéficas.
As mutações que não afectam de forma sensível a capacidade reprodutiva (como as mutações de expressão tardia) podem encontrar-se em vários membros da família desde longa data, sendo herdadas através das células germinais de geração em geração. Nos indivíduos com origem num ovo portador de mutação, todas as células nucleadas do organismo são portadoras das mutações (incluindo as da linha germinal), denominando-se esta condição como constitucional.
Ocasionalmente, uma mutação pode ocorrer de novo num membro de uma família, a nível dos gâmetas, do zigoto, das primeiras fases do desenvolvimento embrionário, em etapas mais avançadas do desenvolvimento embrionário ou fetal, ou mesmo após o nascimento. Se os gâmetas forem portadores desta mutação e esta der origem a uma anomalia ou a uma doença, sem afectar significativamente a capacidade reprodutiva, inicia-se assim a instalação na família em causa, já referida. No entanto, este tipo de mutação pode ser um letal genético, isto é, pode não permitir a reprodução, embora sem afectar a sobrevivência do seu portador (o que, em termos genéricos, equivale à morte daquele genoma, dada a impossibilidade de se reproduzir). Um raciocínio idêntico pode ser feito em relação às mutações letais nos primeiros anos de vida. Nestes dois casos, o aparecimento de novos casos da doença apenas se verifica por mutação de novo, sendo a frequência da doença aproximadamente igual à taxa de mutação do gene em causa, se não houver mortalidade pré-natal associada à mutação.
Quando uma mutação ocorre numa fase do desenvolvimento ontogénico posterior à determinação celular que origina as células germinais e não atinge estas células, a mutação não se transmite à descendência. É uma mutação somática que, na família em questão, afecta apenas o indivíduo em causa e origina uma eventual condição esporádica de doença (por exemplo, algumas mutações somáticas responsáveis por casos esporádicos de cancro).
As mutações somáticas podem ser letais e podem permitir a divisão da célula ou mesmo a sua morte, instalando-se no genoma das células filhas. Apesar de haverem mecanismos de reparação de lesões do DNA nas células, quando a quantidade de mutações é grande ou quando ocorrem em organismos nos quais há deficiências para proceder a essas reparações, as alterações não são reparadas e instalam-se nas células descendentes.
Cromossomopatias
As cromossomopatias dizem respeito a fenótipos patológicos determinados por alterações cromossómicas estruturais ou numéricas. Associam-se normalmente a atrasos mentais e a malformações congénitas múltiplas. Quando em conjunto, são responsáveis por cerca de metade dos abortos espontâneos, ocorrendo estes em 20% das gravidezes conhecidas. Entre as alterações cromossómicas que se encontram mais vulgarmente nos abortos espontâneos contam-se as trissomias (em 50% dos casos) e as monossomias do X (em 20%). A prevalência de cromossopatias nos recém-nascidos ronda os 0,6%.
As cromossopatias dividem-se em heterossómicas (quando dizem respeito a alterações do cromossoma X ou do Y) e em autossómicas (quando ocorrem em algum dos cromossomas dos pares 1 a 22). Destas últimas as mais comuns em recém-nascidos são a trissomia 21 (síndroma de Down), a trissomia 18 (síndroma de Edwards) e a trissomia 13 (síndroma de Patau). As cromossopatias heterossómicas mais comuns são a monossomia do X (síndroma de Turner), a trissomia XXY (síndroma de Klinefelter), a trissomia XXX (síndroma da super-fêmea) e a trissomia XYY (síndroma do super-macho). Todas estas resultam da não-disjunção meiótica, na primeira ou na segunda divisão, e serão explicadas mais à frente.

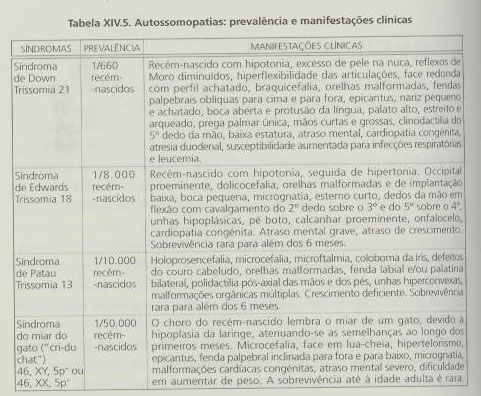
O número de cromossomas X supranumerários ou em falta nos gâmetas do sexo feminino é independente do facto de a não-disjunção ocorrer na primeira ou na segunda divisão da meiose. Contudo, quando a não-disjunção tem lugar na primeira divisão, os dois cromossomas X presentes em cada gâmeta feminino são portadores das diferenças que se encontram em cromossomas homólogos, em comparação com a identidade completa dos cromossomas X quando a não-disjunção ocorre na segunda divisão. Neste caso, os dois cromossomas do gâmeta feminino resultam de um par de cromátideos.

No caso do sexo masculino as consequências são diferentes, dependendo da ocorrência das não-disjunção na primeira ou na segunda divisão meiótica, no que respeita aos cromossomas X ou Y supranumerários ou em falta. Quando ocorre na primeira divisão, originam-se gâmetas nulissómicos e gâmetas XY. Da fecundação com um oócito normal podem resultar embriões com trissomia XXY ou com monossomia X. Caso a não-disjunção ocorra na segunda divisão meiótica, originar-se-ão gâmetas YY e XX que, após fecundarem os oócitos normais, darão origem a embriões com trissomia XYY ou com trissomia XXX.
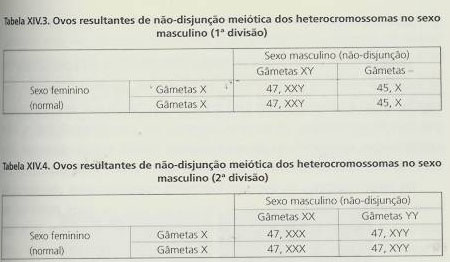
Trissomia 21 (Síndroma de Down)
Esta aneuploidia foi descrita em 1866, é a mais frequente na população, com uma prevalência de aproximadamente 1/660 recém-nascidos que é atingida em mulheres com gravidezes a termo a partir dos 31 anos, aumentando de forma progressiva e muito significativa com o avanço da idade materna. Não são verificáveis diferenças de incidência significativas quando comparadas diferentes raças humanas, grupos sociais e regiões geográficas.
Cerca de 0,5% dos embriões têm trissomia 21, contudo a sua incidência é maior no momento da fecundação do que ao nascer, estimando-se que cerca de 70% dos embriões abortem espontaneamente.
Sendo este síndroma uma condição sintomática com possibilidade de se expressar por uma multiplicidade de anomalias, é necessário salientar que nenhuma das anomalias físicas está presente em 100% dos doentes, que algumas das características podem mesmo encontrar-se em indivíduos normais e que as mesmas características se podem observar em diversas patologias com organização sintomática diversa.
Há então a salientar vários aspectos:- Os recém nascidos apresentam, como manifestação mais frequente, hipotonia muscular, laxidão articular e excesso de pele na zona posterior do pescoço (em 80% dos casos).
- A pele tem um aspecto característico denominado por cutis marmorata, podendo verificar-se cianose dos dedos e dos lábios em relação com a existência de cardiopatia.
- A cabeça é oval e pequena, evidenciando braquicefalia (alongamento do diâmetro bi-parietal), e achatamento occipital, as orelhas são de tamanho reduzido, com lobos pequenos ou mesmo ausentes.
- A face é arredondada, com perfil achatado (em 90% dos casos), as fendas palpebrais são oblíquas para cima e para fora (em 80% dos casos), sendo frequente a prega no epicantus.
- Na íris observa-se um ponteado de cor esbranquiçada por falta de pigmentação (manchas de Brushfield).
- O nariz é pequeno e achatado.
- A boca está geralmente aberta e é corrente a protrusão da língua.
- Há hipoplasia do maxilar inferior e o palato é estreito, alto e muito arqueado.
- As mãos são curtas e grossas, com dedos também curtos, observando-se, em 60% dos doentes, hipoplasia da falange média do 5º dedo da mão e, por vezes, clinodactilia deste mesmo dedo.
- Têm, em 45% dos casos, uma prega palmar única uni ou bilateral (situação que ocorre em 2 a 5% dos indivíduos normais).
- Os pés são curtos e verifica-se um aumento do espaço entre o 1º e o 2º dedos.
- Há displasia da pélvis em 70% dos casos e, por vezes, atrésia duodenal.
- Em 40 a 60% dos afectados encontra-se a cardiopatia congénita.
- O atraso mental é uma das manifestações major nesta trissomia, estando presente em todos os doentes (o QI médio de um adulto doente é, geralmente, de 24).
- A voz é gutural e apresentam dificuldade na articulação das palavras.
- O crescimento das crianças é lento, o que origina baixa estatura e maturação óssea retardada.
- O estrabismo encontra-se em 20% dos indivíduos, bem como outras anomalias oculares (como, por exemplo, glaucoma e cataratas).
- Têm grande susceptibilidade para as infecções respiratórias.
- O risco de desenvolverem leucemia aguda é de aproximadamente 1% (10 a 20 vezes superior à prevalência na população geral, sendo a leucemia megacariocítica aguda a mais frequente).
- Os que vivem para além dos 40 anos desenvolvem quase sempre a doença de Alzheimer.
- A esperança média de vida é mais curta, dependendo a sobrevivência da gravidade das lesões e da possibilidade de as reparar cirurgicamente, nomeadamente a nível cardíaco.
 Com o conhecimento do genoma (em particular do cromossoma 21), das funções e do modo de actuação das proteínas codificadas pelos genes localizados no cromossoma em questão, é possível antever formas de intervenção que possam contrariar as deficiências de desenvolvimento, como sejam o atraso mental. Com o conhecimento do genoma (em particular do cromossoma 21), das funções e do modo de actuação das proteínas codificadas pelos genes localizados no cromossoma em questão, é possível antever formas de intervenção que possam contrariar as deficiências de desenvolvimento, como sejam o atraso mental.
A confirmação do diagnóstico implica sempre um estudo citogenético. Aproximadamente 93 a 95% dos casos de trissomia 21 (por vezes também designado mongolismo) devem-se à presença de um cromossoma 21 supranumerário num dos gâmetas (trissomia livre) originado pela não-disjunção meiótica (cariótipo 47,XX+21 ou 47,XY+21).
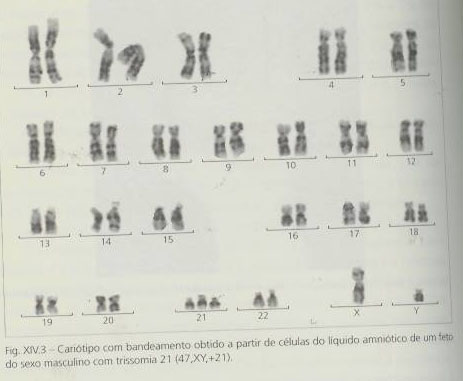
Este cromossoma supranumerário, em 90% dos casos, é de origem materna, ocorrendo a não-disjunção na primeira divisão meiótica (em 75% das vezes, em associação com a diminuição de uma taxa de recombinação meiótica). Nos restantes casos, ocorre na segunda divisão, não parecendo existir relação coma taxa de recombinação meiótica. A idade materna influência significativamente a ocorrência da não-disjunção.
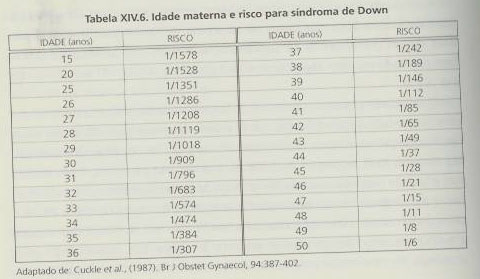
O cromossoma supranumerário tem origem na não-disjunção meiótica paterna em apenas 5 a 7% dos casos. Anomalias durante a mitose são ainda responsáveis pela existência deste mesmo cromossoma excedentário em cerca de 3 a 5% dos casos. Também as translocações robertsonianas são responsáveis por 3 a 4% das situações, bem como as translocações de novo em 2 a 3% (quase sempre de origem materna e, geralmente, originadas antes do crossing-over da primeira divisão) e, em 1 a 2%, as translocações herdadas de um dos progenitores em que se encontram de uma forma equilibrada. Estas ocorrem normalmente entre um cromossoma 21 e um cromossoma 13,14 ou 15, ou ainda entre os dois cromossomas 21. A idade da mãe não tem qualquer tipo de influência na ocorrência da translocação.
O mosaicismo, responsável por cerca de 2,5% dos casos, é um acontecimento pós-zigótico (cariótipo 47,XY+21/46,XY ou 47,XX+21/46,XX). A origem mais comum deste é a perda do cromossoma 21 supranumerário em algumas células de um embrião inicialmente trissómico, ou à não-disjunção na mitose de um embrião que, inicialmente, era cromossomicamente normal. Nestes casos as manifestações da trissomia são clinicamente mais frustes do que nos casos de outra natureza, dada a presença de células trissómicas e normais simultaneamente. Aqui, a maior ou menor gravidade depende da percentagem de células trissómicas, que se interliga com o momento do desenvolvimento embrionário em que ocorreu a não-disjunção mitótica. Pode eventualmente acontecer que, se o mosaicismo for tardio, apenas se verifique em alguns tecidos.
Trissomia 13 (Síndroma de Patau)
Esta trissomia foi descrita em 1960 e tem uma prevalência de cerca de 1/10000 recém-nascidos, sendo que no período intra-uterino a mortalidade é muito elevada. Uma ecografia fetal cuidadosa pode evidenciar anomalias do cérebro, dos membros (polidactilia), cardiopatias congénitas, anomalias da face (malformações do nariz, fendas, anoftalmia ou sinoftalmia), malformações renais (hipoplasia, hidronefrose) e onfalocelo. O desenvolvimento até à idade adulta é muito raro e só cerca de 3% dos indivíduos é que sobrevivem para além dos seis meses de vida.
Os aspectos fenotípicos mais característicos dos recém-nascidos são:- Microcefalia;
- Lábio leporino (em 2/3 dos casos) geralmente acompanhado de fenda esfenopalatina;
- Polidactilia pós-axial (dos dedos dos pés ou das mãos);
- Anomalias cardíacas e/ou renais;
- Defeitos do couro cabeludo;
- Em 66% dos casos está presente a malformação mais grave da trissomia 13, que é como quem diz a holoprosencefalia ;
- Entre as malformações severas do sistema nervoso central pode encontrar-se ainda hipoplasia do cerebelo, ausência de tractos e bolbos olfactivos (arrinencefalia), hidrocefalia e agenesia do corpo caloso;
- Cegueira;
- Surdez;
- Convulsões epiléticas;
- Dificuldades na alimentação;
- Alargamento da sutura sagital e das fontanelas;
- Orelhas malformadas de implantação baixas e com apêndices auriculares e a displasia retiniana;
- Criptorquidia no sexo masculino;
- Malformações renais, gastrintestinais e cardíacas;
- Hangiomas na pele, sobretudo na fronte;
- Em 60% dos casos há uma prega palmar única, e os dedos estão em flexão permanente.
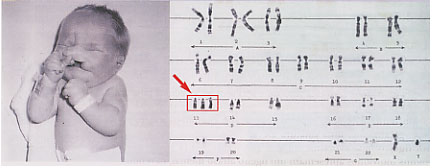
Em aproximadamente ¾ dos casos verifica-se a existência de trissomia livre, tendo o cromossoma supranumerário origem na não-disjunção meiótica (normalmente na mãe 90% dos casos). As translocações robertsonianas são responsáveis por cerca de 20% dos casos. Metade dos casos de translocação é herdada (da mãe) e a outra metade resulta de mutações de novo.
Para uma mulher jovem com um filho com trissomia 13 livre, o risco de recorrência é de aproximadamente 1%, sendo que para mulheres de idade mais avançada deverá ser adicionado o risco de 1%, o risco decorrente da idade materna, estando indicada a realização de estudo citogenético pré-natal.
Trissomia 18 (Síndroma de Edwards)
Este tipo de trissomia, descoberto em 1960, ocupa o segundo lugar em termos de frequência das malformações múltiplas mais comuns. Tem uma prevalência de 1/8000 recém-nascidos, representando, no entanto, este valor apenas 5% dos fetos, já que 95% deles são abortados espontaneamente. Verifica-se também que a frequência é três vezes maior nas raparigas do que nos rapazes. Devido à gravidade, à multiplicidade das malformações e à incapacidade para se desenvolverem, poucos são os que sobrevivem para além do ano de idade, morrendo metade durante a primeira semana de vida. Os que conseguem sobreviver para além do ano de idade correspondem a crianças com atraso mental grave e com incapacidade para andar sem apoios, sendo apenas 5 a 10% dos casos.
Durante a gravidez, é vulgar detectar-se polihidrâmnios, placenta pequena e, por vezes, uma artéria umbilical única, havendo geralmente atraso de crescimento intra-uterino, movimentos fetais hipocinéticos e podem ser detectadas malformações. Nos recém-nascidos pode encontrar-se:- Atrasos do crescimento;
- Hipotonia muscular seguida de hipertonia após o período neonatal;
- Resposta fraca aos estímulos sonoros e choro fraco;
- Episódios de apneia;
- Fraca capacidade de sucção (o que pode obrigar ao recurso a uma sonda nasogástrica para proceder à alimentação);
- Pele redundante;
- Occipital proeminente;
- Sutura metópica aberta;
- Dolicocefalia (redução do diâmetro biparietal);
- Malformação das orelhas, de implantação baixa;
- Fendas palpebrais curtas;
- Boca pequena com lábio superior curto e palato muito arqueado;
- Os dedos das mãos encontram-se em flexão com cavalgamento do 2º sobre o 3º e do 5º sobre o 4º, com unhas hipoplásicas, podendo haver sindactilia;
- Calcanhar proeminente;
- Pescoço fino;
- Esterno curto e com um reduzido número de pontos de ossificação;
- Costelas e clavículas hipoplásicas, por vezes, com fragmentação;
- Na parede abdominal podem observar-se eventrações ou hérnias inguinais ou umbilicais e diastasi recti devido a defeitos da musculatura;
- Criptorquidia nos órgãos genitais masculinos e hipertrofia do clitóris e dos grandes lábios no sexo feminino, apresentando também ovários hipoplásicos;
- Cardiopatias congénitas, sobretudo septais;
- Malformações gastrintestinais (pâncreas ectópico, má rotação do cólon);
- Segmentação anormal dos pulmões;
- Rim ectópico e rim em ferradura;
- Atraso mental.

A trissomia 18 é causada, maioritariamente, pela presença de um cromossoma 18 supranumerário resultante da não-disjunção meiótica. Também o avanço da idade materna está associado à maior incidência desta patologia na descendência.
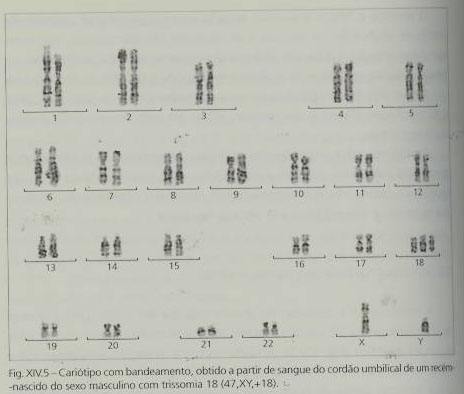
A translocação e o mosaicismo são também uma possível causa etiológica, embora rara. Nos casos de mosaicismo, a gravidade das implicações fenotípicas depende da percentagem de células trissómicas. Também pode ocorrer a trissomia parcial do braço curto do cromossoma 18 (neste caso as consequências fenotípicas não são específicas e o atraso mental pode ser moderado ou inexistente) e a trissomia parcial do braço longo do mesmo cromossoma (com as mesmas características da trissomia livre ou então incaracterísticas).
Síndroma de Turner
Esta síndroma foi descrita em 1938, em indivíduos do sexo feminino tendo como alterações características a baixa estatura, infantilismo sexual e amenorreia primária. Tem uma prevalência de 1/2500 recém-nascidos do sexo feminino e a sua causa mais frequente é a monossomia do X, correspondente a um cariótipo 45,X. Os embriões e os fetos afectados têm uma viabilidade muito reduzida devido ao aborto espontâneo, estimando-se que menos de 1% sobrevivam até ao parto. nos casos em que o aborto é mais precoce, é mais provável a presença de uma monossomia em que o cromossoma X presente é de origem paterna, enquanto que nos abortos tardios (do segundo trimestre da gravidez) é mais provável encontrar um cromossoma X de origem materna.
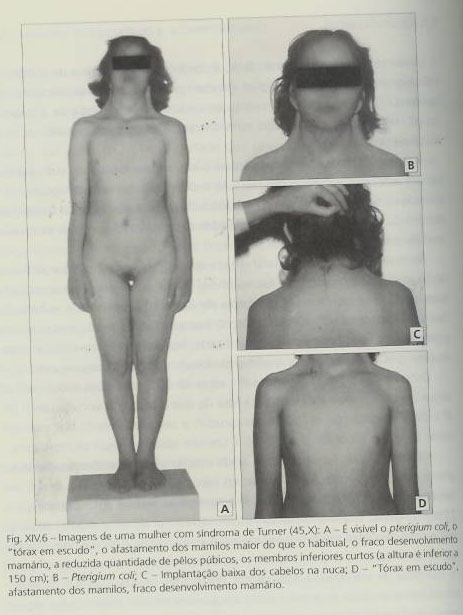
Relativamente aos aspectos clínicos é de salientar que:- É frequente, durante o desenvolvimento fetal, a ocorrência de hidrópsia e de higromas quísticos na região cervical, por excesso de acumulação de líquido. O pterigium coli (pele redundante a sugerir asas, no ângulo do pescoço com os ombros), está relacionado com a existência de higroma.
- Ao nascer, observa-se um pescoço curto, implantação baixa do cabelo (em 2/3 dos casos), orelhas de implantação baixa, linfedema das mãos, dos pés, dos dedos dos pés, o que acarreta hipoplasia das unhas, e verifica-se também pterigium coli.
- No desenvolvimento pós-natal, regista-se atrasos no crescimento e ausência de pulo de crescimento na adolescência, o que leva a uma acentuada baixa da estatura, com uma altura média entre os 130 e os 140 centímetros, devida sobretudo a membros inferiores curtos.
- Em cerca de 50% dos casos, verifica-se cubitus valgus (antebraço mais inclinado para fora do que o braço), o encurtamento do 4º e do 5º metacarpos, o tórax em escudo, um afastamento maior dos mamilos do que o esperado e nevos pigmentados.
- Podem ocorrer algumas anomalias viscerais graves como a coarctação da aorta (em aproximadamente 10% dos casos), o rim em ferradura e duplicação ureteral.
- Em 27% dos casos está presente a hipertensão arterial, havendo também uma maior incidência de otite média, tiroidite autoimune, doença de Crohn, diabetes mellitus na idade adulta e hemorragias gastrintestinais.
- O grau de inteligência está dentro dos limites normais, apesar de se verificar um défice quando se procede à comparação com irmãos.
- Pode ocorrer algum atraso na fala e dificuldades de aprendizagem.
- Observa-se uma perturbação da percepção espacial e o comportamento social é frequentemente afectado, de modo significativo, quando o cromossoma X presente é de origem materna. Quando este é de origem paterna, o comportamento social é muito semelhante ao de uma mulher normal.
- Há amenorreia primária (ausência de menarca), esterilidade, falta de desenvolvimento dos caracteres sexuais secundários e níveis séricos de gonadotrofinas elevados.
- Em algumas mulheres com este síndroma, sobretudo quando está presente mosaicismo 45,X/46,XX, podem-se observar menarca e menstruações durante alguns meses ou anos, sendo a menopausa de ocorrência precoce. Ocasionalmente registaram-se gravidezes.
Em cerca de 55% dos casos de síndroma de Turner está presente uma monossomia do cromossoma X, correspondente a um cariótipo 45,X. Neste caso a idade materna não tem qualquer tipo de relação com a não-disjunção envolvida nesta síndroma. Em 80% dos casos, o cromossoma presente tem origem materna, pelo que a causa não será a não-disjunção meiótica paterna.
Para além da monossomia 45,X este síndroma pode também dever-se a mosaicismo 45,X/46,XX, a mosaicismo 45,X/46,XY (em 5% dos casos), a isocromossoma do braço longo do cromossoma X, a cromossoma X em anel, e ainda a outras alterações estruturais como deleções de um dos braços do cromossoma X. As manifestações fenotípicas nos casos de isocromossoma para o braço longo do cromossoma X são idênticas às que se observam em mulheres 45,X.
A probabilidade de recorrência de síndroma de Turner é muito reduzida.
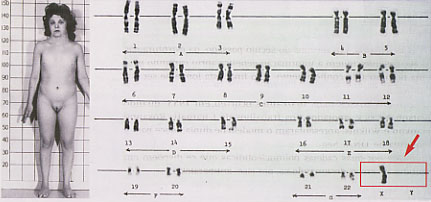
Síndroma de Klinefelter
Esta síndroma é uma aneuploidia, descrita pela primeira vez em 1942, com uma prevalência de cerca de 1/600 recém-nascidos do sexo masculino. Resulta da presença de um cromossoma X supranumerário num indivíduo com fenótipo masculino, devido à não-disjunção num dos progenitores. Esta pode ter lugar na mãe ou no pai, ocorrendo com proporções aproximadamente iguais. Sabe-se que a idade das mães tem alguma relação, mas não de forma tão acentuada como para as demais trissomias.
Convém ainda referir que nas polissomias do X mais extensas do que a que ocorre desta síndroma (por exemplo, 48,XXXY; 49,XXXXY), à medida que o número de cromossomas X aumenta, se vai acentuando a redução da capacidade intelectual e surgem também malformações cardiovasculares, ósseas e da face.
Como aspectos clínicos que alertam para o diagnóstico da síndroma de Klinefelter salientam-se:- Hipogonadismo;
- Microorquidia;
- Esterilidade por azoospermia;
- Ginecomastia (30% dos casos);
- Elevação dos valores séricos das gonadotrofinas FSH e LH e redução dos níveis de testosterona;
- Ao nascer, os testículos aparentam um tamanho e uma consistência normais e, pelo menos até à puberdade, podem-se encontrar células da linha espermatogénica. No adulto, a regra é a presença de microrquidia traduzida num comprimento dos testículos entre 1 a 2 centímetros, comparativamente com 3,5 a 4,5 centímetros nos indivíduos normais.
- Observam-se alterações nos túbulos seminíferos;
- Elevada estatura, devido sobretudo ao comprimento dos membros inferiores;
- Em 25% dos casos está presente a clinodactilia.
- Não há atraso mental, embora a inteligência tenda a ser mais reduzida que a dos irmãos;
- Apresentam dificuldades na aprendizagem, especialmente na leitura;
- Há uma maior susceptibilidade para problemas comportamentais em condições de stress e para a depressão;
- Têm uma auto-imagem deficiente, sentem-se frustrados com facilidade, e a capacidade para a relação interpessoal é fraca;
- Têm normalmente um fraco crescimento da barba;
- O pénis é de tamanho normal, geralmente;
- A libido está diminuída, embora possa haver erecções, coito e ejaculação;
- Normalmente são estéreis;
- Nos indivíduos adultos há um risco acrescido para o cancro da mama, para tumores de células germinais, para a osteoporose e para as doenças autoimunes.
A trissomia 47,XXY está presente em cerca de 85% dos casos de síndroma de Klinefelter. O mosaicismo 46,XY/47,XXY ocupa quase por inteiro os restantes 15% dos casos, embora tenham sido encontrados outros mosaicos, mais raros, como o 46,XX/47,XXY ou o 46,XX/46,XY/47,XXY.
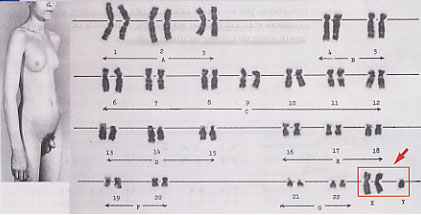
Trissomia XXX
Esta trissomia, designada por vezes como síndroma da super-fêmea ou da super-feminilidade, tem uma prevalência de aproximadamente 1/1000 recém-nascidos do sexo feminino, sendo que a maioria dos casos tem origem na não-disjunção materna, verificando-se também aqui um aumento da incidência com o avanço da idade.
As manifestações fenotípicas não são muito aparentes, podendo observar-se anomalias esqueléticas e hipertelorismo. Embora possa haver um ligeiro défice comparativo quando comparados com os irmãos, a inteligência destes indivíduos está dentro dos padrões normais. Há muitas vezes problemas com a linguagem verbal, havendo também, com frequência, necessidade de apoios educativos acrescidos. A transmissão de um cromossoma supranumerário à descendência é improvável e a fertilidade não é afectada.
Trissomia XYY
Esta trissomia, denominada vulgarmente por síndroma da supermasculinidade ou do super-macho, tem uma prevalência de cerca de 1/1000 recém-nascidos do sexo masculino, resultando o cromossoma Y supranumerário da não-disjunção na meiose paterna.
Normalmente não há manifestações significativas a nível do fenótipo dos indivíduos afectados, sendo a estatura maior do que a média para a população o dado mais saliente. Os seus portadores tendem a apresentar fraco desenvolvimento da musculatura peitoral e da cintura escapular e uma deficiente coordenação motora dos movimentos finos. Na adolescência têm acne nodulocístico severo, e podem apresentar o tamanho dos dentes aumentado. Tal como nas últimas cromossopatias referidas, a inteligência está dentro dos parâmetros normais (a não ser quando comparada com a dos irmãos que é ligeiramente superior), têm dificuldades de aprendizagem principalmente a nível da linguagem. O comportamento agressivo não costuma ser um problema habitual.
Os seus portadores são maioritariamente férteis, não havendo risco significativo de ter filhos com esta anomalia. Pode, ocasionalmente, observar-se criptorquidia, redução do tamanho do pénis ou hipospadias.
|
|

